Running nf-core pipelines
Software requirements for nf-core pipelines
In order to run nf-core pipelines, you need to have Nextflow installed (https://www.nextflow.io). The only other requirement is a software packaging tool: Conda, Docker or Singularity. On Genotoul, we use Singularity.
Usage instructions and documentation
Each pipeline has its own webpage at https://nf-co.re/PIPELINE.
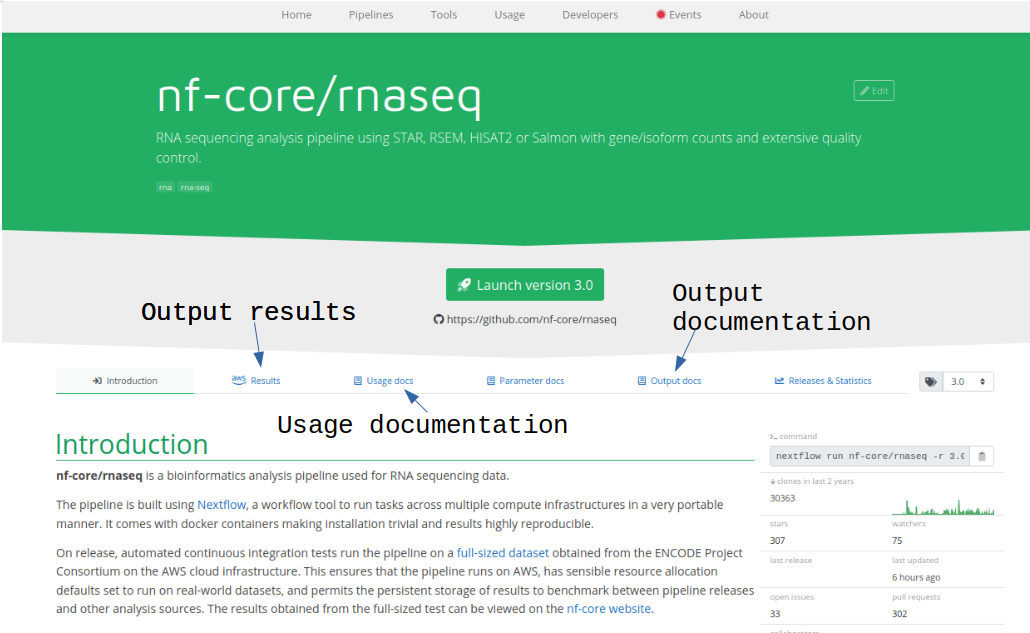 See online documentation for rnaseq pipeline
See online documentation for rnaseq pipeline
In addition to this documentation, each pipeline comes with basic command line reference. This can be seen by running the pipeline with the --help flag, for example:
$ nextflow run nf-core/rnaseq --help
General informations
Genomes with nf-core:
- We do not provide igenome
- You can use genome from
/bankingenologin - As we are not sure about software used to generate indexes we recommand to
- Run one time workflow with
--fastaand--gtf(or more if required) and--save_reference - Indexes will be available in
results/genome - Create a
genome.configfile and use it while rerun analysis with--genome GenomeName. More info in genome
- Run one time workflow with