Nextflow config
There are different ways to configure nextflow execution : with configuration file or with profiles (defined in configuration).
- With config file
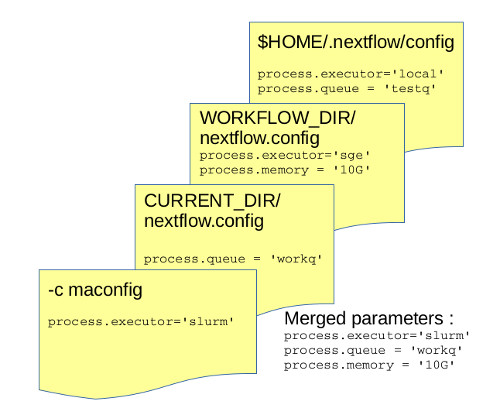
Nextflow looks for config in following order and only new params are loaded (ie the settings in the first override the same ones that may appear in the second one)
- in current directory (eg file created previously
nextflow.config) - in workflow directory (eg
~/.nextflow/assets/nextflow-io/rnaseq/nextflow.config) - in home directory
$HOME/.nextflow/config - finally if config file is given with
-c <config file>, the settings are considered as the first.
If you want to ignore any default configuration files and use only the custom one use the command line option
-C <config file>.
Configuration file example:
#scope by dot prefixing
process.executor = 'slurm'
process.queue = 'workq'
process.memory = '10G'
#scope using the curly brackets
singularity {
enabled = true
autoMount = true
}
- With profiles
Configuration files can contain the definition of one or more profiles. A profile is a set of configuration attributes that can be activated/chosen when launching a pipeline execution by using the -profile command line option.
profiles {
standard {
process.executor = 'local'
}
cluster {
process.executor = 'slurm'
process.queue = 'unlimitq'
process.memory = '10GB'
}
Exercice 5:
- View the config use by nextflow-io/hello, by looking at the file nextflow.config which is store in workflow dir (
nextflow info) - Use the command the
nextflow configto also see the config (merged with current config) - Rename the file
nextflow.configin current directory bynextflow.config.save - Re-execute the nextflow config command.